{kind=link}
![]()
![]()
Physical maps from linked reads — rewritten in Rust.
A ground-up rewrite of Physlr for constructing de novo physical maps
from 10x Genomics Chromium or MGI stLFR linked-read data.
- Overview
- Installation
- Quick Start
- Pipeline Details
- Commands
- Parameters
- Reproducing Results
- Project Structure
- Citation
Physlr 2 takes linked-read sequencing data and constructs ordered physical maps — sets of molecules arranged along each chromosome. These maps can then scaffold draft genome assemblies to chromosome-level contiguity.
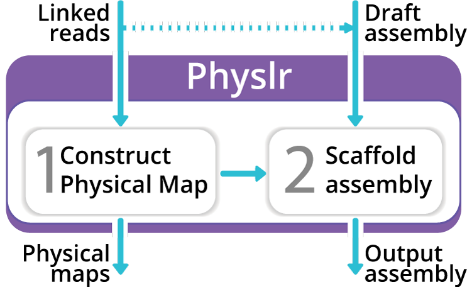
Two main tasks:
- Physical map construction — builds an ordered map of molecules along each chromosome from linked-read barcodes.
- Assembly scaffolding — uses the physical map to order and orient contigs from a draft genome assembly.
Optional: merge-paths — a post-processing step that identifies non-backbone "bridge" molecules sharing minimizers with endpoints of adjacent backbone paths, providing evidence to merge them. On human stLFR data (NA24143), this adds 9 true-positive merges with zero false positives.
Backbone paths mapped to the GRCh38 reference genome (NA24143, stLFR):
| Backbone | Reference |
|---|---|
 |
 |
Backbone view: paths colored by reference chromosome. Reference view: chromosomes colored by backbone path. Click to enlarge.
curl --proto "=https" --tlsv1.2 -sSf https://sh.rustup.rs | sh
source ~/.cargo/envgit clone https://github.com/aafshinfard/physlr2.git
cd physlr2
cargo build --releaseChoose one:
# Option A: Add to PATH (recommended)
export PATH="$(pwd)/target/release:$PATH"
# Option B: Install to ~/.cargo/bin
cargo install --path .
# Option C: System-wide
sudo cp target/release/physlr /usr/local/bin/physlr --version
physlr --help| Dependency | Purpose | Install |
|---|---|---|
| btllib | Repeat filtering and minimizer extraction (indexlr, ntcard, nthits) |
conda install -c bioconda btllib |
| Dependency | Purpose | Install |
|---|---|---|
| Python 3 + matplotlib | Backbone-vs-reference visualization | pip install matplotlib |
| Snakemake ≥ 7 | Automated workflow | conda install -c bioconda snakemake |
| QUAST | Reference-based assembly evaluation | conda install -c bioconda quast |
Physlr provides three commands, from most automated to most granular:
Reads + draft assembly → physical map + scaffolds, all in one command.
physlr pipeline reads.fq.gz --draft draft.fa -o output/Output:
| File | Description |
|---|---|
physlr.backbone.path |
Physical map (backbone paths) |
physlr.filtered.tsv |
Filtered barcode minimizers |
physlr.scaffolds.fa |
Scaffolded assembly |
physlr.report.json |
Before/after assembly metrics |
Add -g for NG50 reporting (optional):
physlr pipeline reads.fq.gz --draft draft.fa -o output/ -g 3088269832Reads → physical map, without scaffolding.
physlr physical-map reads.fq.gz -o output/Uses an existing physical map (from physical-map) to scaffold a draft assembly.
physlr scaffolds output/physlr.backbone.path output/physlr.filtered.tsv draft.fa -o output/Arguments:
- Backbone path file (from
physical-map) - Filtered minimizer TSV (from
physical-map) - Draft assembly FASTA
For full control over each stage:
# 1. Build repeat Bloom filter (ntcard → nthits → physlr-makebf)
physlr repeat-filter reads.fq.gz -o repeats.bf -k 32 -w 32 -t 16
# 2. Index minimizers with repeat filtering (uses indexlr from btllib)
physlr index reads.fq.gz -o reads.mxs.tsv -k 32 -w 32 --repeat-bf repeats.bf
# 3. Filter barcodes and minimizers
physlr filter-minimizers reads.mxs.tsv -o filtered.mxs.tsv -n 100 -N 5000
# 4. Compute barcode overlap graph
physlr overlap filtered.mxs.tsv -o overlap.tsv
# 5. Filter edges by percentile
physlr filter-overlap overlap.tsv -o overlap.filtered.tsv -p 85
# 6. Separate barcodes into molecules
physlr molecules overlap.filtered.tsv -o mol.tsv --strategy bc+cc
# 7. Extract backbone paths (physical map)
physlr backbone mol.tsv -o backbone.path
# 8. (Optional) Merge adjacent backbone paths
physlr split-minimizers mol.tsv filtered.mxs.tsv -o split.mxs.tsv
physlr merge-paths backbone.path split.mxs.tsv -o merged.path
# 9. (Optional) Scaffold a draft assembly
physlr index-contigs draft.fa -o draft.mxs.tsv
physlr map backbone.path filtered.mxs.tsv draft.mxs.tsv -o map.bed
physlr bed-to-path map.bed -o scaffold.path
physlr path-to-fasta draft.fa scaffold.path -o scaffolds.faLinked reads (FASTQ + barcodes)
│
├── repeat-filter Build repeat Bloom filter (ntcard → nthits → physlr-makebf)
├── index Extract (k,w)-minimizers per barcode (indexlr from btllib)
├── filter-minimizers Remove low/high-count barcodes, singleton minimizers
├── overlap Compute barcode overlap graph (shared minimizers)
├── filter-overlap Remove low-weight edges by percentile
├── molecules Separate barcodes into individual molecules
├── backbone MST → prune branches → extract paths
│
└──► Physical map (backbone paths)
│
├── merge-paths (Optional) Merge adjacent paths via bridge molecules
├── map / map-paf Map contigs or reference to the physical map
├── bed-to-path Convert mappings to scaffold paths
├── path-to-fasta Produce scaffolded FASTA
│
└──► Scaffolded assembly
| Command | Description |
|---|---|
pipeline |
End-to-end: FASTQ → physical map → scaffolded assembly |
physical-map |
Build a physical map from linked-read FASTQ files |
scaffolds |
Scaffold a draft assembly using an existing physical map |
| Command | Description |
|---|---|
index |
Extract (k,w)-minimizers from FASTA/FASTQ, grouped by barcode |
index-contigs |
Extract ordered minimizers from FASTA contigs or reference |
repeat-filter |
Detect repetitive k-mers and build a Bloom filter |
| Command | Description |
|---|---|
filter-minimizers |
Filter barcodes by count; remove singleton/repetitive minimizers |
overlap |
Compute barcode overlap graph from shared minimizers |
filter-overlap |
Remove low-weight edges by percentile |
| Command | Description |
|---|---|
molecules |
Separate barcodes into individual molecules |
split-minimizers |
Assign barcode minimizers to individual molecules |
trace-molecules |
Diagnostic: trace molecule separation for specific barcodes |
| Command | Description |
|---|---|
backbone |
Extract backbone paths from the molecule overlap graph |
merge-paths |
Merge adjacent backbone paths using bridge molecule evidence |
| Command | Description |
|---|---|
map |
Map query sequences to the physical map (BED output) |
map-paf |
Map sequences to the physical map (PAF output, for visualization) |
bed-to-path |
Convert BED mappings to scaffold paths |
path-to-fasta |
Produce scaffolded FASTA from scaffold paths |
| Command | Description |
|---|---|
metrics |
Compute assembly metrics (N50, NG50, etc.) |
path-metrics |
Compute physical map metrics |
backbone-dot |
Generate DOT visualization of backbone paths |
Most parameters have sensible defaults. The tables below are for advanced tuning.
| Parameter | Default | Description |
|---|---|---|
-k |
32 | K-mer size for minimizer extraction |
-w |
32 | Window size for minimizer extraction |
-t |
auto | Number of threads (auto-detected, capped at 16) |
-v |
1 | Verbosity: 0 = silent, 1 = info, 2 = debug |
Controls which barcodes and edges are kept in the overlap graph.
| Parameter | Default | Description |
|---|---|---|
-n / --min-count |
100 | Min minimizers per barcode (removes low-coverage barcodes) |
-N / --max-count |
5000 | Max minimizers per barcode (removes chimeric/noisy barcodes) |
--min-shared |
10 | Min shared minimizers to create an overlap edge |
-p / --percentile |
90 | Remove the bottom N% of edges by weight (~85 for stLFR, ~92.5 for 10x) |
Controls how the minimum spanning tree is pruned to extract backbone paths.
| Parameter | Default | Description |
|---|---|---|
--prune-branches |
10 | Remove branches shorter than this from MST junctions |
--prune-bridges |
10 | Remove bridge edges connecting components smaller than this |
--prune-junctions |
200 | Remove junction branches shorter than this |
--min-component-size |
50 | Discard backbone paths shorter than this (in molecules) |
Merges adjacent backbone paths using bridge molecule evidence. Defaults optimized on human stLFR data for zero false positives.
| Parameter | Default | Description |
|---|---|---|
--endpoint-depth |
25 | Molecules from each path end used as endpoints |
--min-endpoint-hits |
4 | Bridge must connect ≥ N endpoint molecules per side |
--min-bridges |
2 | Min bridge molecules to accept a merge |
--min-shared-mx |
3 | Min shared minimizers between bridge and endpoint |
--max-connections |
2 | Discard bridges connecting > N paths (specificity) |
--max-links-per-endpoint |
1 | Discard endpoints with > N candidate links (ambiguity) |
--min-bridge-density |
0.01 | Min ratio of bridges to shorter path length |
| Parameter | Default | Description |
|---|---|---|
-n / --min-score |
10 | Min mapping score for contig-to-physical-map mapping |
--gap-size |
100 | Ns inserted between scaffolded contigs |
-g / --genome-size |
— | Expected genome size in bp (optional, for NG50 only) |
See REPRODUCING.md for step-by-step instructions to reproduce the NA24143 results shown above, including data download links and the automated pipeline script.
physlr2/
├── Cargo.toml # Package manifest
├── src/
│ ├── main.rs # CLI entry point (clap)
│ ├── lib.rs # Library root
│ ├── minimizer/mod.rs # Minimizer extraction and filtering
│ ├── overlap/mod.rs # Barcode overlap computation
│ ├── molecules/mod.rs # Molecule separation
│ ├── graph/mod.rs # Graph algorithms (MST, pruning)
│ ├── backbone/mod.rs # Backbone extraction + merge-paths
│ ├── map/mod.rs # Mapping to physical map
│ ├── scaffold/mod.rs # Assembly scaffolding
│ ├── repeat/mod.rs # Repeat k-mer detection (Bloom filter)
│ ├── report/mod.rs # Metrics and reporting
│ └── io/mod.rs # File I/O (TSV, FASTA, BED, gzip)
├── scripts/
│ ├── plotpaf.py # Backbone-vs-reference visualization
│ ├── find-ntcard-mode.py # K-mer histogram mode finder
│ └── profile_pipeline.sh # Pipeline profiling (time + memory)
├── workflow/
│ ├── Snakefile # Snakemake pipeline
│ └── scripts/ # Workflow helper scripts
├── results/ # Example result plots
├── REPRODUCING.md # Reproducibility instructions
└── LICENSE # GPL-3.0
If you use Physlr, please cite:
Afshinfard, A., Jackman, S.D., Wong, J., Coombe, L., Nikolic, V., Chu, J., Mohamadi, H., & Birol, I. (2022). Physlr: Next-Generation Physical Maps. DNA, 2(2), 116–130. doi:10.3390/dna2020009