Need help for branch mismatch problem #1
Description
Hello Dr TimCoorens
Thanks to you for creating the build_phylogeny algorithm, I am using this wonderful algorithm well for work.
During running this algorithm, there was a problem that the branch number and the actual sample in the input file did not match.
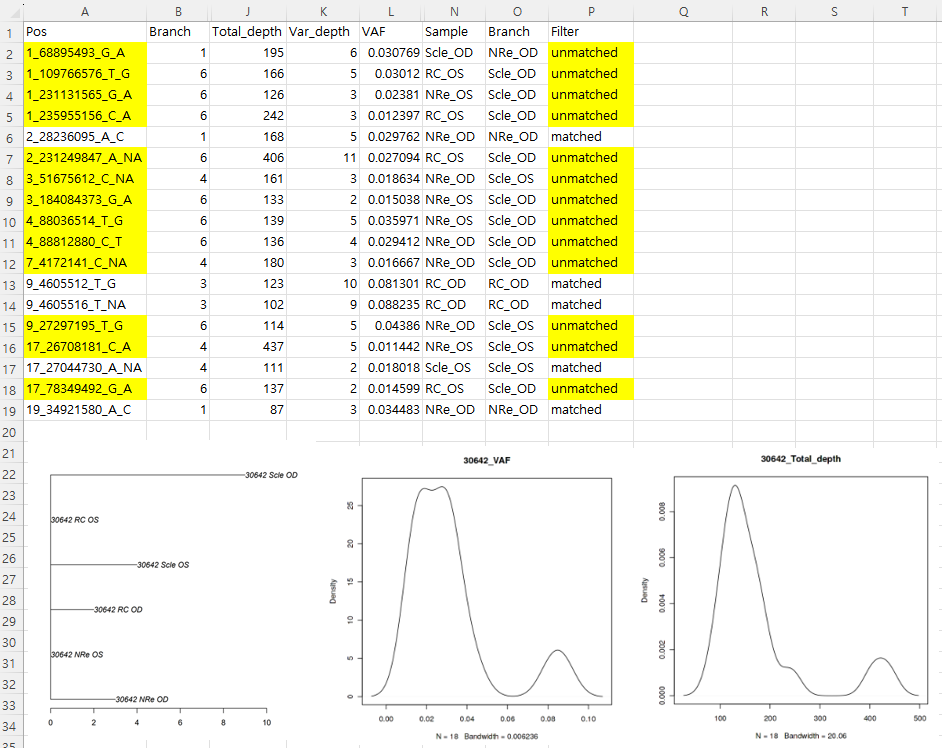
I checked the cases, and found that most of the cases were with variants that have low VAF & high read depth.
So, I ask you if there's a problem already known in low VAF condition (or high depth) and how can I handle it.
Could you give me a solution?
my situation is this:
I used 6 samples of 1 patient. Those samples are not micro-dissectioned, so not clonal as guessed.
When I used the option--vaf_present 0.3 --vaf_absent 0.1, each sample of all the variants inassigned to treefile matched well with the branch number. However, when I used the option--vaf_present 0.1 --vaf_absent 0.01, I found there are not a few variants that have a branch mismatch problem.
I included a summarized xlsx file for easy understanding in the zip file below. Also all the input files, intermediate products, and output tree files for detailed investigation.